


阿立哌唑对亚洲双相I型障碍急性躁狂或混合发作患者的疗效和安全性:安慰剂对照,双盲试验研究(AMAZE研究)(拔萃)
摘要
目的:调查阿立哌唑用于亚洲双相I型障碍躁狂或混合发作患者的疗效和安全性。
方法:该多中心、双盲研究将受试者随机分至阿立哌唑(24mg/d,如果不耐受降至12mg/d)组(n=128)或安慰剂组(n=130),治疗3周。主要疗效评估YMRS总分自基线的平均变化。
结果:总计136例患者(阿立哌唑组56.3%,安慰剂组49.2%)完成该研究。大多数患者(92.6%)接受24mg/d阿立哌唑治疗。自第4天至第3周,阿立哌唑较安慰剂显著改善YMRS总分(-11.3vs.-5.3,P<0.001)。最常见不良事件(>15%的患者,阿立哌唑vs.安慰剂)为静坐不能(22.0%vs.5.6%)和失眠(16.3vs.9.6%)。阿立哌唑治疗组自基线对体重的影响与安慰剂组无显著差异(-0.4vs.-0.7kg,P=0.231)o阿立哌唑不增加受试者的泌乳素水平。
结论:阿立哌唑治疗亚洲双相I型障碍急性躁狂或混合发作患者的疗效显著优于安慰剂,且阿立哌唑应用安全,耐受性好。
序言
急性双相躁狂的主要治疗目标是快速改善症状。非典型抗精神病药物也被大多数指南推荐用于双相I型障碍相关急性躁狂症状的一线治疗,阿立哌唑为其中之一。阿立哌唑对多巴胺D2和D3受体发挥部分激动作用,对5-羟色胺5-HT2A发挥拮抗作用,对5-HT1A起到部分激动作用。
研究方法
在日本、中国香港、台湾、大陆,印尼,马来西亚和菲律宾共计91个中心进行此随机双盲、安慰剂对照试验。试验分为两个期,7天的筛选期和3周的双盲治疗期。所有患者都被随机接受阿立哌唑24mg/d或安慰剂治疗3周,阿立哌唑起始剂量24mg/d,如果不耐受可将剂量降至12mg/do
結果
总计342例患者纳入此研究,其中258例被随机分组分至阿立哌唑治疗组(11=128)或安慰剂组(n=130),阿立哌唑治疗组72例(56.3%)、安慰剂组64例(49.2%)完成试验。
研究期内阿立哌唑的平均剂量为22.9mg/d,大多数患者(92.6%)的患者接受阿立哌唑24mg/d,6.6%接受12mg/d,0.8%接受18mg/d治疗。
1.疗效
第3周,阿立哌唑较安慰剂显著改善YMRS总分(-11.3vs.-5.3,P<0.001),见图。阿立哌唑组患者的YMRS总分自第4天改善较安慰剂组更显著(-6.4vs.-2.9,P<0.001)。
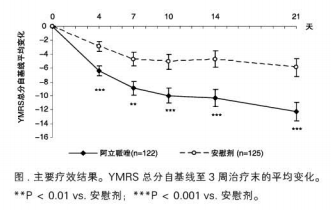
主要次级终点,阿立哌唑治疗组患者的CGI-BP(双相障碍临床总体印象量表)疾病(躁狂)严重度评分自第4天直至研究终点的改善均较安慰剂组更显著(-0.6vs.-0.3,P<0.001)。
阿立哌唑组和安慰剂组由于缺乏疗效导致的停药率分别为23.0%和28.8%(优势比1.4;95%CI0.8-2.4,P=0.289)。
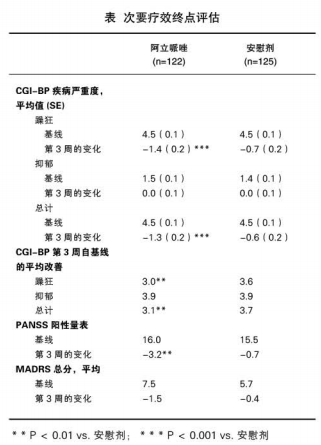
阿立哌唑组在一些其它疗效评估中也显示出自基线改善显著,见表。阿立哌唑组在第3周改善CGI-BP疾病严重度评分更显著。阿立哌唑治疗组第3周的CGI-BP躁狂和总分自基线均较安慰剂组改善显著(P<0.01),阿立哌唑治疗组第3周的PANSS阳性量表自基线较安慰剂组也改善显著(P<0.01),阿立哌唾组的第3周的MADRS评分自基线改善较安慰剂组明显。
2.安全性和耐受性
体重
两组患者研究终点的体重自基线均轻度下降(阿立哌唑-0.4kg,安慰剂-0.7kg,P=0.231)。
两组自基线至3周研究期间的的辛普森-安格斯量表(SAS)和异常不自主运动量表(AIMS)平均变化之间无显著差异(阿立哌唑vs.安慰剂,SAS:0.2vs.-0.1,P=0.163;AIMS:-0.1vs.-0.1,P=0.703)。
泌乳素水平
双盲治疗期间,阿立哌唑治疗组的平均血清泌乳素水平降低653111IU/L,安慰剂组下降497111IU/L,P=0.070。
生命体征等
生命体征变化或其它实验室分析结果显示两组间无临床显著差异。无患者显著增加总胆固醇,甘油三酯或血糖水平。
该研究为迄今为止第一个评估非典型抗精神病药物治疗东南亚双相I型障碍患者的获益的大型、随机、安慰剂对照试验。试验结果显示,阿立哌唑显著改善亚洲双相I型障碍躁狂或混合发作患者的躁狂症状,且耐受性好。
医生投稿:文章来源于医生投稿,内容仅代表作者个人观点,不代表本平台立场,转载请联系原作者。
 举报
举报 点赞
点赞 分享
分享





