


抗癌反促癌?Cell重磅发布麻省理工&哈佛团队突破性研究:肝细胞过度适应环境,或是致癌前奏
 学术前沿官方号
学术前沿官方号 关注
关注肝脏是我们身体最重要的代谢与解毒器官,但在长期高脂饮食、肥胖、糖尿病等代谢压力下,肝细胞不得不应对营养过剩和毒性环境。在肝细胞癌(HCC)的临床研究中,存在一个几乎被默认接受的认知,即肿瘤的出现,源于长期慢性肝病背景下突变的逐步积累。代谢功能障碍相关脂肪性肝病(MASLD)、病毒性肝炎或酒精性肝病,被视为提供“土壤”的背景因素,而真正推动肝癌发生的关键节点,往往被归因于基因层面的失控[1]。
然而,随着代谢相关肝癌发病率在全球范围内持续上升,这一线性模型正面临越来越多的挑战。临床实践中并不难发现,在相似的代谢负荷、炎症程度甚至纤维化阶段下,只有一部分患者最终发展为肝癌。这提示我们,在基因突变发生之前,环境压力已可通过表观遗传重塑和细胞状态改变,为肿瘤发生“铺路”[2]。
近期,来自麻省理工学院(MIT)与哈佛医学院及其附属麻省总医院的联合研究团队,在权威期刊《细胞》(Cell)上发表了一项系统性研究,首次从单细胞和多组学层面,完整描绘了肝细胞在长期慢性代谢应激下的适应性演变轨迹。
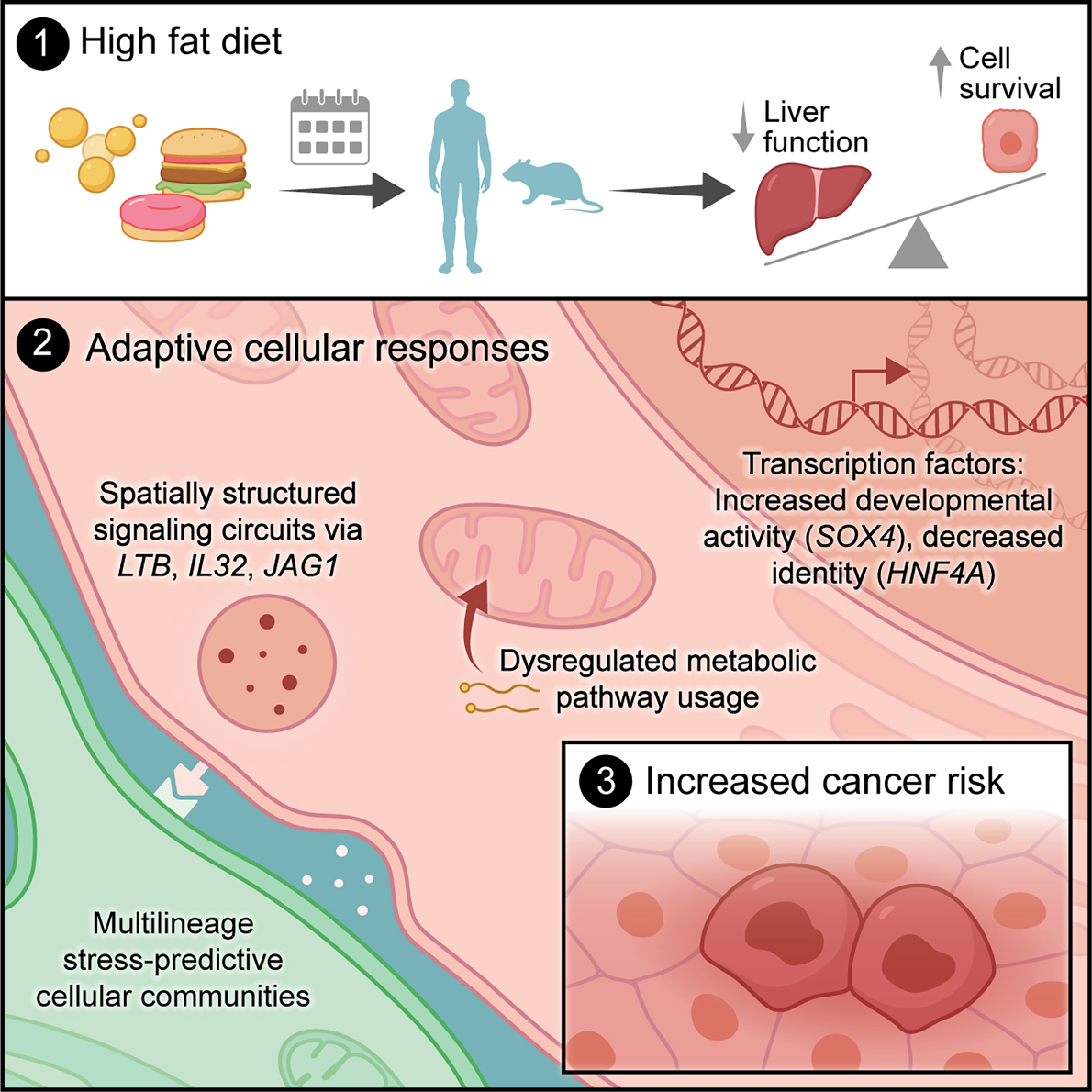
图1 环境压力下的肝细胞求生模式,为何会悄然提高未来癌变风险[3]
研究设计:不依赖致癌物,只观察真实的慢性代谢压力[3]
这项研究最大的特点,在于其模型设计高度贴近真实世界的代谢性肝病进程。研究团队并未引入任何化学致癌物或遗传工程操作,而是仅通过长期高脂饮食(HFD),在小鼠体内诱导出从脂肪肝、炎症、纤维化直至自发性肝细胞癌的完整演变过程。
在这一时间跨度极长的模型中,研究者对肝组织进行了多层级、纵向的系统分析,包括单细胞转录组测序、单细胞染色质可及性分析、蛋白质组学及脂质组学,并进一步结合人类MASLD与HCC队列进行验证。
这种设计使研究不再局限于肿瘤发生后发生了什么,而是首次系统性回答了一个更本质的问题,也就是在肿瘤出现之前,肝细胞究竟已经发生了哪些根本性改变。
核心发现一:慢性代谢应激下,肝细胞正在悄然改变[3]
研究首先发现,在长期代谢负荷的作用下,肝细胞并非被动受损,而是主动启动了一系列适应性反应。这些反应在短期内可能具有保护意义,但在长期持续存在时,却逐渐改变了肝细胞的基本身份。
通过对不同时间点肝细胞转录特征的系统分析,研究者发现,一部分基因表达程序随时间持续增强,主要与细胞存活、应激耐受和再生相关;而另一部分与成熟肝细胞功能密切相关的程序,则呈现出持续下降趋势。
换言之,肝细胞正在逐渐放弃其高度分化的代谢与合成功能,转而优先维持“活下去”的能力。这种状态并非典型的去分化,而是一种介于成熟肝细胞与再生样状态之间的“功能让位型适应”。
值得注意的是,这种变化并非发生在肿瘤组织中,而是广泛存在于尚未发生癌变的肝实质细胞中。
核心发现二:癌前阶段已经确定,肿瘤只是顺势登场[3]
进一步分析显示,这些在慢性代谢应激下形成的转录程序,在随后形成的肝癌组织中被进一步放大。也就是说,肿瘤细胞并没有创造全新的异常状态,而是继承并强化了早期已经存在的应激适应程序。
这一发现直接挑战了既往肿瘤发生主要由突变驱动的传统线性模型。研究提示,在突变真正发挥致癌作用之前,肝细胞已经被长期代谢压力塑造成一种更容易被推向肿瘤的状态。
从这个角度看,肝癌更像是一种长期错误适应的最终结果,而非一次偶然事件。
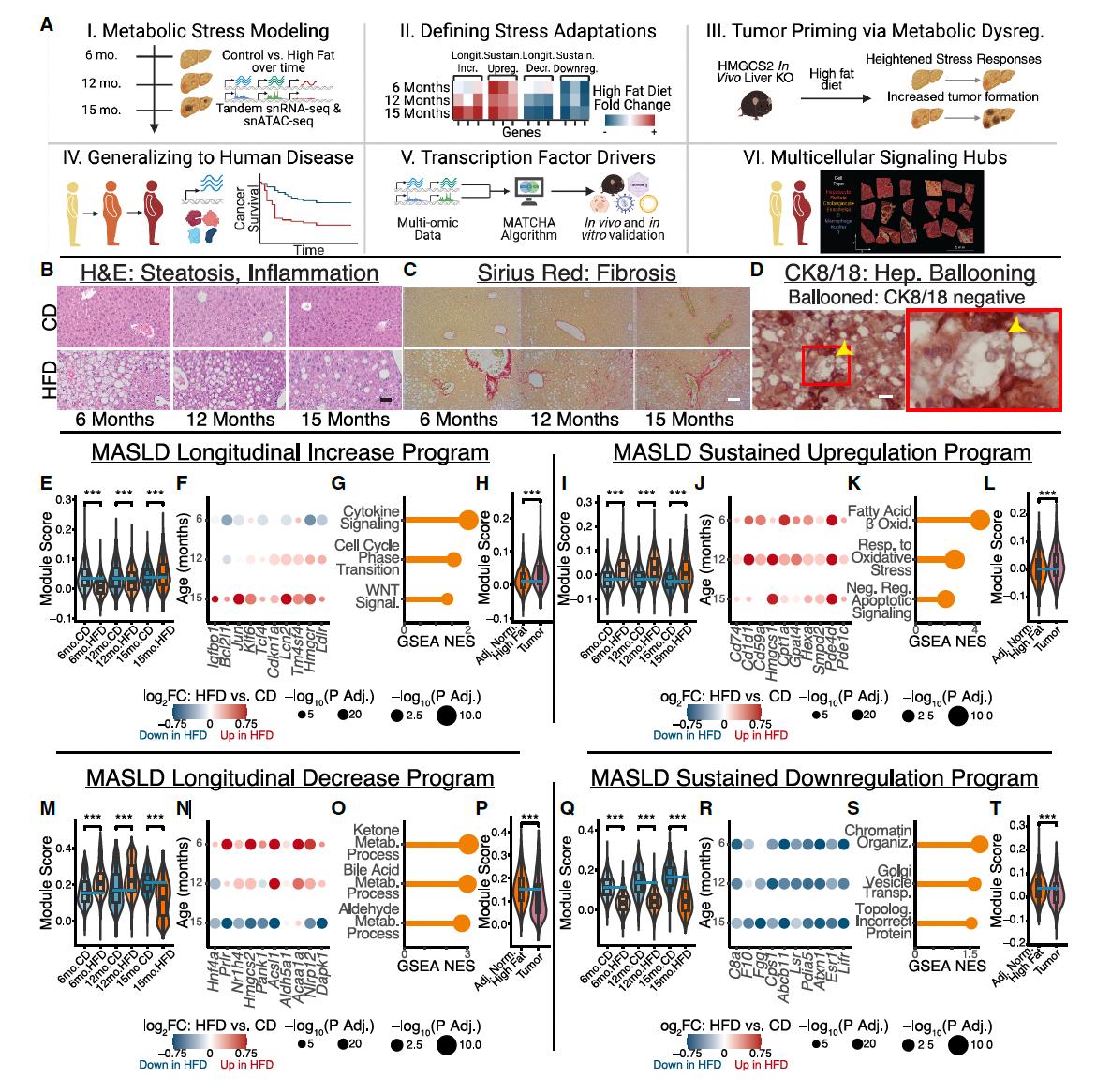
图2 肝细胞在慢性代谢应激下的动态反应[3]
核心发现三:关键代谢节点HMGCS2持续下调改变会结局走向[3]
在所有发生显著变化的基因中,研究者特别关注了HMGCS2(一种线粒体型HMG-CoA合成酶)。HMGCS2在代谢压力下持续下调,导致酮体生成减少。酮体是脂肪酸氧化的终产物,其减少意味着脂质代谢中间体(如乙酰辅酶A)积累,可能促进表观遗传改变和细胞增殖信号。
研究团队发现,HMGCS2在慢性代谢应激过程中持续下调,这一变化不仅出现在小鼠模型中,在人类HCC队列中同样存在。更重要的是,在非肿瘤肝组织中,HMGCS2的低表达与未来肝癌发生风险显著相关。
功能实验进一步证实,在肝细胞中缺失 HMGCS2,会加重代谢应激反应,推动促存活、促增殖程序的激活,并显著增加肿瘤发生倾向。这一结果提示,代谢路径从酮体生成向胆固醇及生物合成通路的偏移,本身就具有促癌预设效应。
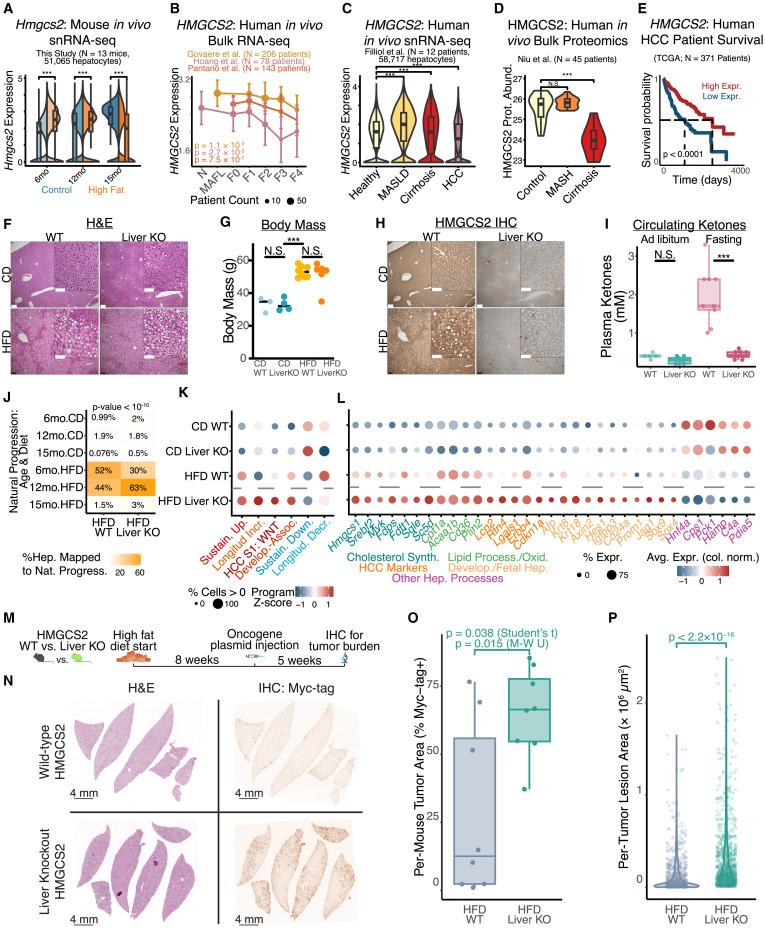
图3 HMGCS2作为肝细胞应激反应和肿瘤启动调节因子的体内验证[3]
核心发现四:表观遗传层面的提前“松动” [3]
除了转录层面的改变,研究还揭示了一个更早发生的事件——染色质可及性的重塑。
通过单细胞ATAC-seq分析,研究者发现,在肿瘤尚未形成时,与肿瘤相关信号通路(如 WNT信号)相连的染色质区域已经变得更加开放。这种变化先于基因表达的明显上调,提示肝细胞在表观遗传层面已经完成了“预热”。
这意味着,当后续突变或信号刺激出现时,肝细胞更容易迅速进入促增殖和促肿瘤状态。肿瘤的发生,不再是突然启动,而是顺着早已被铺好的轨道加速前进。
值得注意的是,研究发现这些慢性应激诱导的转录和表观遗传程序,并非仅存在于代谢性肝病相关肝癌中。在病毒性肝炎和其他肝损伤模型中,也可观察到高度相似的变化趋势。
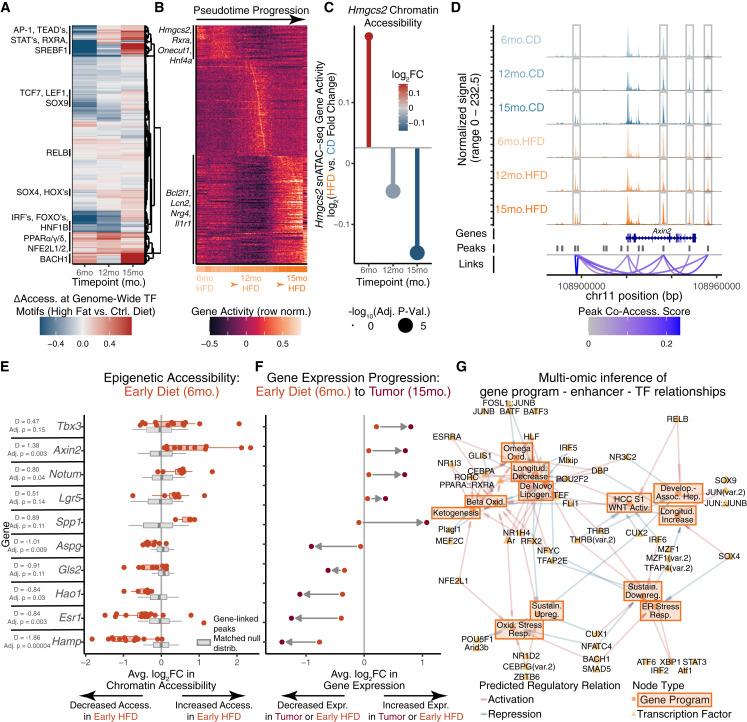
图4 慢性代谢应激驱动表观遗传轨迹改变和WNT通路启动[3]
核心发现五:SOX4与RELB是推动失控适应的分子开关[3]
在多组学整合分析中,研究团队识别出多个在慢性代谢应激中起核心调控作用的转录因子,其中SOX4和RELB尤为突出。
研究显示,这些转录因子在代谢应激过程中逐步上调,能够同时增强促存活、促再生信号,并抑制成熟肝细胞功能基因的表达。在体内实验中,SOX4的过表达足以推动肝细胞更快进入高度应激和增殖状态。
在人类MASLD和HCC样本中,这些分子的表达变化与疾病进展和预后密切相关,进一步印证了其临床相关性。
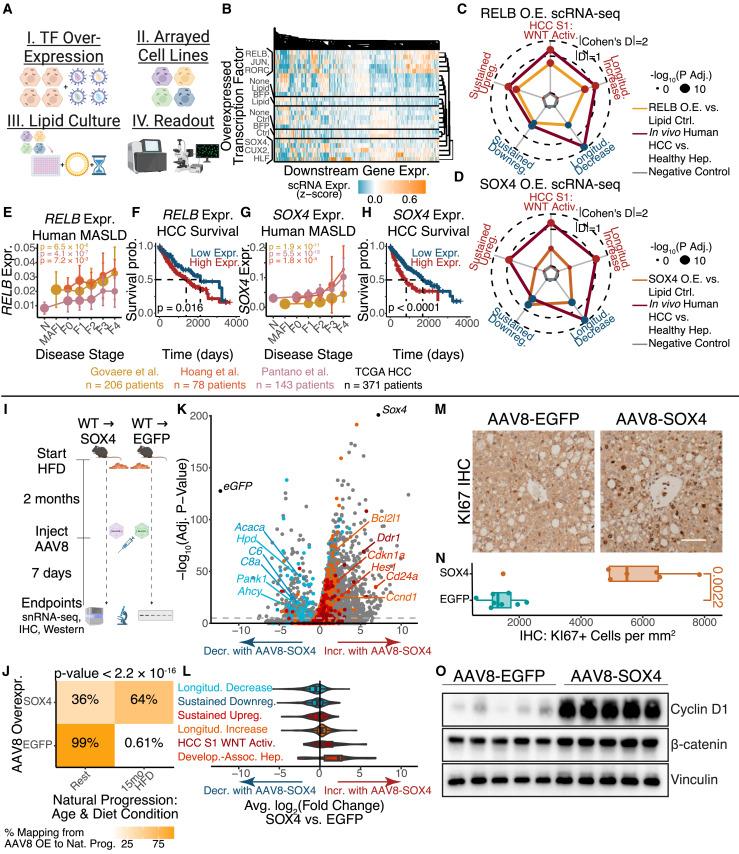
图5 OX4与RELB在肝细胞代谢应激反应中的调控作用:体外人源实验与小鼠体内遗传干扰验证[3]
总 结
这项研究并非孤立存在。近年来,越来越多的研究支持炎症记忆和代谢记忆在癌症发生中的重要性。这些研究共同指出,细胞对短期压力具有记忆能力,这种记忆可通过表观遗传方式维持,并在长期影响组织稳态和癌变风险[4,5]。因此,早期干预代谢紊乱、控制炎症,可能有助于减轻这种促癌记忆,降低远期肝癌风险。
总之,这项研究为我们理解肝癌提供了一个全新的视角。肿瘤的发生不再只是突变累积的结果,而是慢性代谢应激驱动下,肝细胞在转录、代谢和表观遗传层面逐步被重塑的必然结局。
从临床角度来看,这意味着真正的干预窗口,可能远早于肿瘤被影像学发现的时间点。如何识别并逆转这种癌前适应状态,或许将成为未来代谢相关肝癌防治的关键方向。
参考文献:
1. Liu Y, Hao C, et al. The Role of Oxidative Stress in the Development and Therapeutic Intervention of Hepatocellular Carcinoma. Curr Cancer Drug Targets. 2023;23(10):792-804.
2. Aiello C, Felli E, et al. Rewriting the MASLD-associated hepatocellular carcinoma script: Targeting epigenetics and metabolism. Int J Cancer. 2025;157(10):1991-2003.
3. Tzouanas CN, Shay JES, et al. Hepatic adaptation to chronic metabolic stress primes tumorigenesis. Cell. Published online December 22, 2025.
4. Del Poggetto E, Ho IL, Balestrieri C, et al. Epithelial memory of inflammation limits tissue damage while promoting pancreatic tumorigenesis. Science. 2021;373(6561):eabj0486.
5. Falvo DJ, Grimont A, Zumbo P, et al. A reversible epigenetic memory of inflammatory injury controls lineage plasticity and tumor initiation in the mouse pancreas. Dev Cell. 2023;58(24):2959-2973.e7.
原创文章:方舟健客版权所有,未经许可不得转载。
 举报
举报 点赞
点赞 分享
分享













